Myotonic Dystrophy Testing & Diagnosis
The path to a myotonic dystrophy (DM) diagnosis can be long and complex. Medical professionals meet patients with DM infrequently and are often not familiar with DM. Symptoms of DM can also mimic more common diseases, which can lead to months – or even years – of medical testing to rule out other potential causes. This can be especially true for those who were the first in their family to be diagnosed. On average, people living with DM1 spend more than six years searching before they are correctly diagnosed, while those living with DM2 spend an average of over 10 years! Once the disorder is suspected, the next step is to confirm the diagnosis with genetic testing.
What an Initial Evaluation for Myotonic Dystrophy May Look Like
Your doctor will ask about your symptoms, and possible symptoms and signs of DM in other family members. Your doctor will also perform a physical examination of you. Sometimes, your doctor can suspect the diagnosis of myotonic dystrophy just based on your symptoms and this physical examination. If there is another family member, who was already diagnosed with DM, it will be easier for your doctor to consider DM. If your doctor suspects DM, the next step is to get genetic confirmation with a blood test.
Electromyography (EMG)
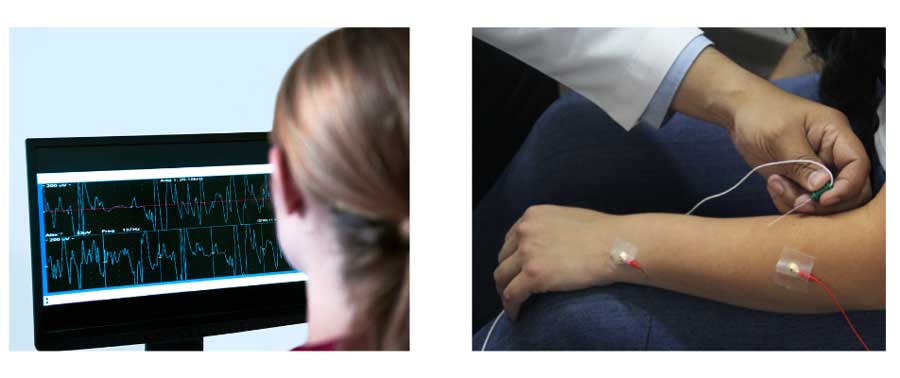
Sometimes your doctor may order additional tests, such as an electromyography (EMG), to evaluate for electrical myotonia (abnormal muscle activity when a small needle is inserted into the muscle). This test can be particularly helpful for patients being evaluated for DM2.
What Genetic Testing for Myotonic Dystrophy Looks Like
A genetic test, also referred to as DNA testing, is required to definitively confirm a diagnosis of DM1 or DM2. The genetic test involves collecting DNA through a blood or saliva sample. The DNA - the genetic material in the nucleus of cells - is then analyzed to determine whether or not you carry the DM1 or DM2 mutation.
Role of a Genetic Counselor: Often times, it is ideal to reach out to a genetic counselor (GC) as early as possible in your journey to a DM diagnosis. The role of a GC encompasses more than genetic test logistics and insurance. The genetic counselor helps you and your family make several important decisions about testing and diagnosis. Conversations with your genetic counselor may cover:
- Understanding the purpose and procedure of a genetic test.
- Deciding amongst a wide range of genetic testing options considering their prices and insurance coverage options.
- Ordering genetic test.
- Discussion about testing and its implications for individuals and families living with DM.
- Family planning with DM.
In general, the genetic counselor works with a doctor to order a genetic test. However, this may differ from state to state based on the licensure laws to order the genetic tests.
Types of genetic tests: Genetic testing for DM1 and/or DM2 uses standard DNA diagnostic protocols:
- PCR (Polymerase Chain Reaction) – which copies segments of DNA found in your saliva or blood.
- Southern Blot – a blood test to confirm the presence of the specific genetic sequence for DM in your DNA.
Costs of these tests are variable and are often dictated by insurance coverage. Generally, commercially available southern blot tests are more expensive than PCR tests. List prices vary from lab to lab, ranging from $250 to $2,000. Your genetic counselor may be able to help navigate pricing of these tests, and may work with your insurance company to try to obtain authorization for testing. Usually, a PCR test is able to provide a yes or no answer regarding a diagnosis of DM1 or DM2, but may not provide an exact repeat number. Your doctor and genetic counselor can inform your decision and recommend the test that is best for you based on your background, family history, and insurance coverage.
Time considerations: The total time from the first visit to a healthcare provider, to the confirmation of the diagnosis via genetic test results, depends on insurance authorization and test turn-around times. Typically, once a sample is sent out for testing, a result is received within 3-4 weeks. The individual can then consult their doctor or genetic counselor to review the results of the test.
Interpreting results: Depending on the test methodology, your report may or may not include an approximate repeat number in the CNBP and/or DMPK gene. Usually, a test will come back with a positive or negative result. In rare scenarios, you may obtain a result with a repeat number in an uncertain or “premutation” range. Your genetic counselor will discuss these results with you upon receipt of the report.
Retesting for DM once a genetic diagnosis is established is not recommended and is unlikely to be covered by insurance.
Benefits of Genetic Testing for Myotonic Dystrophy
A confirmed diagnosis using a genetic test can eliminate the need for additional medical tests and may provide a definitive explanation for many of your symptoms. If you have no symptoms of DM, but other family members carry a diagnosis of DM, genetic testing can help establish whether you carry the DM mutation and therefore are at risk of developing symptoms later in life, or passing DM on to your children.
If you are experiencing symptoms and wonder whether you should seek evaluation for a diagnosis of DM, please consider the following situations in which a diagnosis may help guide or improve your care:
- People living with DM can have trouble with anesthesia. It is helpful for doctors to know about the diagnosis so they can plan ahead accordingly and counsel you better on risks.
- People living with DM are at risk for cardiac abnormalities, in specific cardiac conduction delays. Knowing the diagnosis of DM can help guide cardiac monitoring and care.
- Couples can make family planning decisions. For example, some people with DM may choose to consider in vitro fertilization with pre-implantation diagnostics, which means that embryos are checked for the DM mutation and the embryo without DM is implanted in the women’s uterus.
- Expecting mothers living with DM1 usually receive special monitoring and care during pregnancy, so knowing and sharing that you have DM can help your doctors provide appropriate care.
How Does Repeat Length Relate to the Severity of Myotonic Dystrophy?
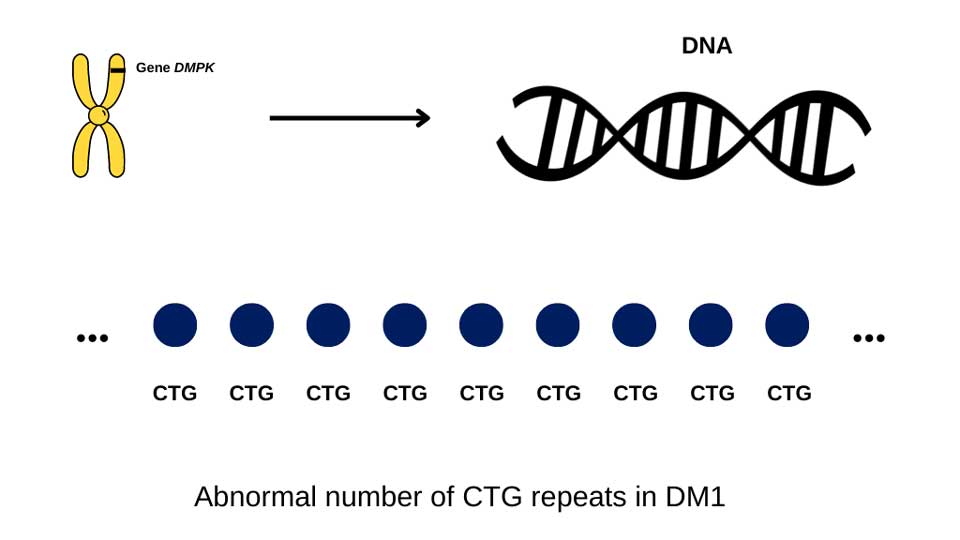
In DM1, the causal mutation is on chromosome 19, where the genetic code (CTG) on a gene called DMPK is expanded. People affected by DM1 have more than 50 CTG repeats, but the number of repeats can range from 50 to more than a thousand when measured in blood.
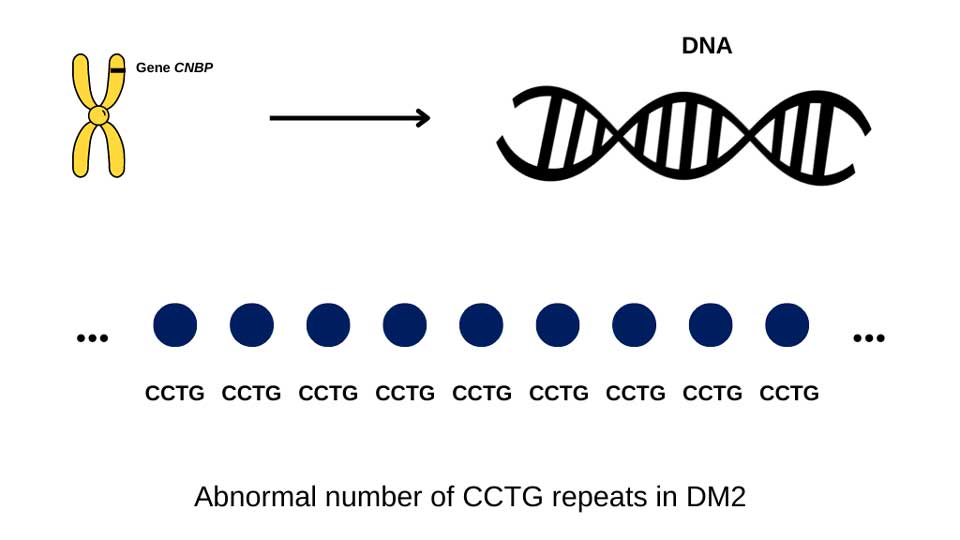
In DM2, the causal mutation is on chromosome 3, where a genetic code (CCTG) on a gene called CNBP is expanded. People with DM2 have more than 75 CCTG repeats, but usually many thousand repeats in blood cells.
In DM1, generally speaking, people who have a low number of CTG repeats (between 50-100), develop symptoms later in life, while those with >1000 repeats may develop symptoms in childhood or may have symptoms at birth. However, in between, above 100 and below 1000 CTG repeats, this connection between number of repeats and symptoms is less clear. For example, if one person has more repeats than another, it does not mean their disease will be more severe.
In DM2, there is no known relationship between CCTG repeat size and symptoms.
For more information on the significance of CTG repeats, watch Dr. Darren Monckton's presentation, Everything You Wanted to Know About CTG Repeats.
Why Does Myotonic Dystrophy Take So Long to Diagnose?
Myotonic dystrophy can cause symptoms affecting multiple organ systems beyond the muscle, e.g. the GI system, eyes, and the heart. You may have seen several different specialists for disparate symptoms, such as an ophthalmologist for blurred vision, a gastroenterologist for stomach pain, diarrhea or constipation, and a cardiologist for an abnormal heartbeat. These individual physicians may not be aware of your full range of problems and therefore may not have put the pieces together for a uniform explanation of your symptoms - myotonic dystrophy.
The severity of symptoms can also vary greatly, even within the same family. People with DM1 can develop symptoms at any point in their lives. Some people with DM1 may have mild symptoms or not show any signs until an older age. Due to this variability, many people may not seek medical attention which can cause their DM to go unrecognized until another family member is diagnosed. DM2 usually affects people later in life, and does not have a congenital form (symptoms at birth).
I Have No Symptoms, Should I Test for Myotonic Dystrophy?
Some people opt against genetic testing when they have no symptoms. Problems that may arise from a definitive diagnosis of DM include:
For more information on the pros and cons of testing, read our interview with Carly Siskind, MS, LCGC, senior genetic counselor on the Stanford University Neuromuscular Disorders Team.
I cannot get evaluated, what can I do? If symptoms of DM and/or a family history of DM exist, but for various reasons it is not possible to get evaluated, the following steps can be taken:
- Consider reaching out to family members to inform them about your condition.
- Continue to seek regular/annual heart screening and share/follow anesthesia guidelines.
- Consider asking your current provider who suspected DM for a referral to, or a remote consultation/satellite care with a neuromuscular specialist.
Where to Test for Myotonic Dystrophy
Individuals pursuing a diagnosis are discouraged from relying on consumer companies focused on ancestry data and genetics for diagnostic results. The sequencing technology used by such companies and their third-party affiliates typically does not have the capacity to produce accurate repeat expansion mutation readings.
There are a number of labs that conduct testing for DM, including academic or commercial laboratories. Clinical (non-research) genetic testing must be ordered by an authorized provider, which can include a doctor, genetic counselor, or both. Work with your care team to discuss lab and testing options that fit your needs. Ask your doctor and/or genetic counselor whether insurance authorization is necessary and what your anticipated cost for testing will be, and whether you may qualify for financial aid or a patient assistance program. Genetic counselors will often take care of any insurance/billing logistics needed prior to testing. However, if you are not working with a genetic counselor you may want to call your insurance company ahead of testing to get an idea of whether the testing will be considered a covered service or not.